Seed Funding
In order to develop and expand innovative ideas, HI-TRON Mainz has so far issued two calls for funding. The "Kick-Start Seed Funding" was published in 2021, followed by the "Seed Funding" call in 2023.
Both calls aim to facilitate the rapid transfer of scientific findings into clinical applications and to exploit synergies between scientists based in Mainz and Heidelberg. The HI-TRON Mainz (Kick-Start) Seed Funding Program therefore provides seed funding for cross-institutional projects with a clear translational focus and a pathway to implementation in patient care. HI-TRON Mainz also supports bringing together basic and clinically active scientists for joint translational (Kick-Start) Seed Funding applications. The proposed projects are highly innovative, align with the focus of HI-TRON and its platforms, and focus on diagnostic and therapeutic aspects of cancer immunotherapy in the areas of "biomarker development" and “identification and validation of novel therapeutic targets.”
Seed Funding 2023
 privat
privat
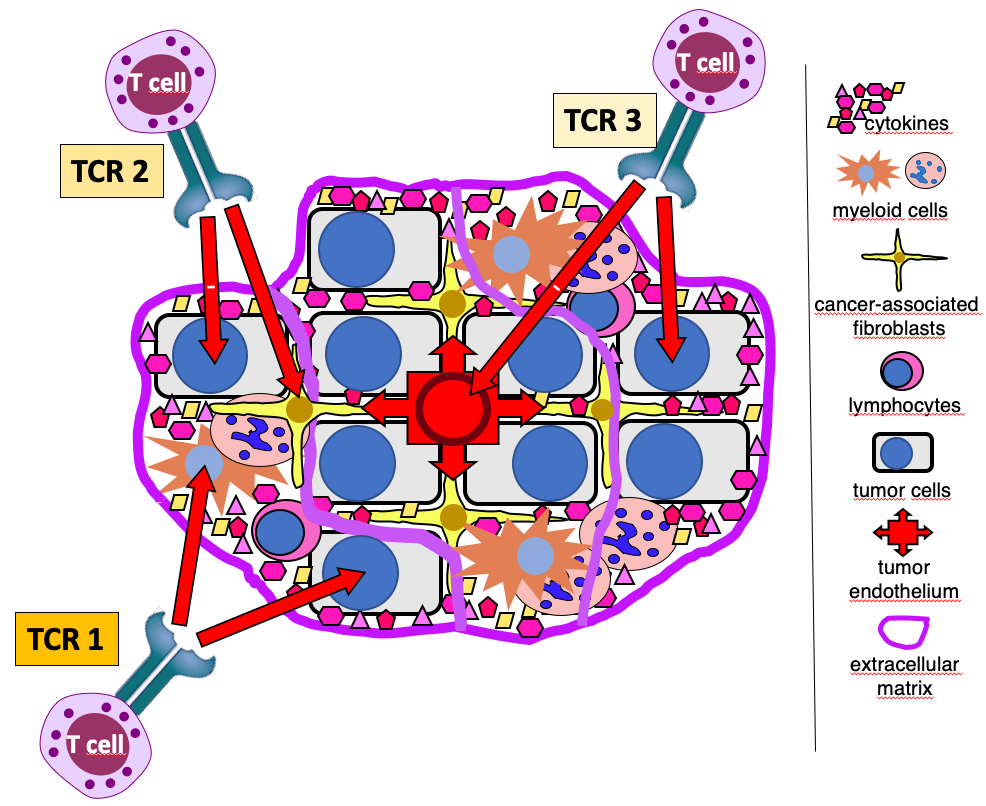
Project 1: Targeting antigens shared by tumor and stromal cells with natural MHC-independent T-cell receptors (Corresponding PI: Thomas Wölfel)
We have identified T-cell receptors (TCRs) that target in an MHC-independent manner distinct antigens that are expressed on tumor cells as well as on stromal cell types forming the tumor microenvironment (TME) that is known to nurture tumor growth and to create barriers for therapeutic interventions of various kinds. The therapeutic use of such TCRs would allow dual targeting of tumor and TME cells. Such, the project addresses two major limitations of cancer immunotherapy. First, the therapeutic use of HLA-independent TCR would counteract down-regulation or loss of HLA expression that represents a frequent immune-escape mechanism in malignant diseases and a serious limitation of T-cell immunotherapy strategies relying on HLA-restricted effectors. Second, targeting at the same time cellular constituents of the TME opens up the opportunity to subvert immunosuppressive features mediated by TME constituents to the point of turning so-called “cold” into “hot” tumors. The aim of the project is to explore the therapeutic window offered by dual-targeting TCRs as well as safety issues associated with their use in pre-clinical mouse models.
Wölfel, Thomas, Prof. Dr. (UM Mainz, internal Medicine III)
Wölfel, Catherine, Dr. (UM Mainz, internal Medicine III)
Echchannaoui, Hakim, Dr. (UM Mainz, internal Medicine III)
Offringa, Rienk, Prof. Dr. (DKFZ, Department D200)
Gaida, Matthias, Prof. Dr. (TRON, Joint Unit Immunopathology)
Volkmar, Michael, Dr. (HI-TRON Mainz, TCR Platform)
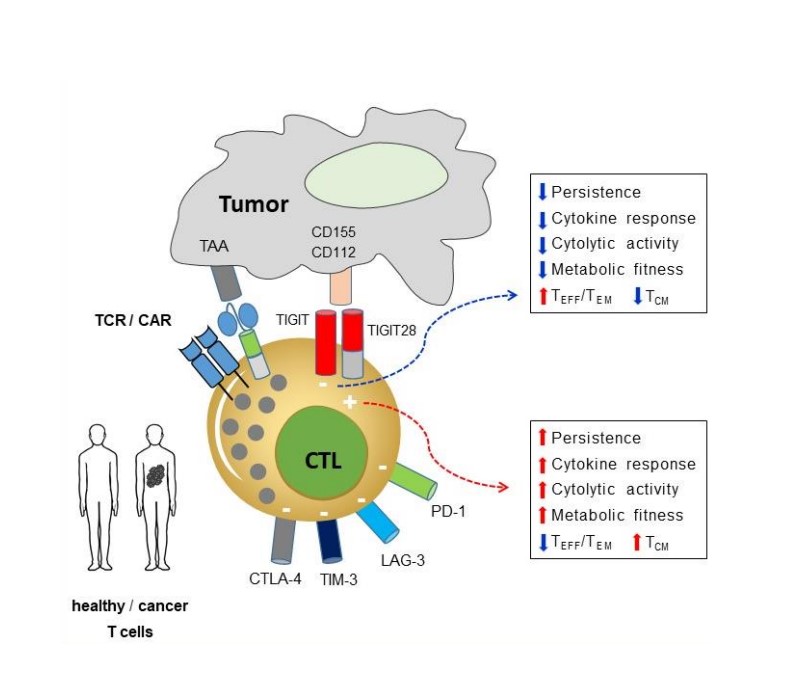
Project 2: Project FIT: Increasing the fitness of CAR/TCR-T cells by co-expressing a novel chimeric switch receptor (Corresponding PI: Hakim Echchannaoui)
Premature exhaustion and ultimately dysfunction of gene modified (CAR/TCR) T cells are often associated with poor therapeutic outcome in cancer patients. Increased expression of inhibitory receptors, namely immune checkpoints (like PD-1, CTLA-4 or TIGIT) are hallmarks of dysfunctional T cells. Immune checkpoint blockers (or ICB) have been developed and widely used in cancer immunotherapy. Despite high numbers of successful immunotherapy trials with ICB, many patients do not respond or show a short-lived response to immunotherapy. A major hurdle remains the resistance and immune-related adverse events to ICB and ultimately the cancer relapse. The project explores an innovative approach to overcome receptor-mediated exhaustion by reprogramming CAR/TCR T cells with fusion proteins containing the extracellular domain of inhibitory and the signaling domain of co-stimulatory receptors (called chimeric switch receptor or SR). Using synthetic biology, we designed a SR targeting the emerging immune checkpoint TIGIT. The prototype SR harbored a CD28 intracellular domain. This concept offers the possibility to convert negative signals from cancer immune checkpoints into activating signals, and thus contributes to reduce or prevent exhaustion and to increase the fitness of anti-tumor T cells. This project is focusing on the functional characterization of the fitness of SR-armed CAR/TCR-T cells in PDX cancer models and 3D bioprinted cultures at the single cell level (transcriptomic, secretomic, metabolic and epigenetic). The data will provide new insights into how optimally designed SR can promote fitness of T cells as a first step towards potential clinical translation.
Dr. phil. nat. Echchannaoui, Hakim (UM Mainz, Department of Internal Medicine III)
Prof. Dr. Halama, Niels (DKFZ, Department of Translational Immunotherapy)
Univ.-Prof. Dr. med. Gaida, Matthias (UM Mainz/TRON Institute for Pathology, Joint Unit Immunopathology)
Univ.-Prof. Dr. med. Theobald, Matthias (UM Mainz, Department of Internal Medicine III)
Dr. med. Legscha, Kevin (UM Mainz, Department of Internal Medicine III)
Antunes, Edite (UM Mainz, Department of Internal Medicine III)
PhD student (UM Mainz, Department of Internal Medicine III)
Research assistant (50%) DKFZ, Department of Translational Immunotherapy)
Kick Start Seed Funding 2021
 G.Cui
G.Cui
Project 1: To enhance anti-tumor immune responses by inhibiting a pair of immune checkpoints (Corresponding PI: Guoliang Cui)
Compared with the traditional cancer therapies, such as chemotherapy and radiation therapy, immune checkpoint blockade-based immunotherapy has higher effectiveness and fewer side effects in several types of cancers. Those immune checkpoints act as ‘brakes’ to suppress the protective immune responses against cancers. Identification and inhibition of those ‘brake’ molecules will unleash anti-cancer immunity. We have identified one of those immune system “brakes” in human and mouse tumors (referred to as “a new IC” in the figure below), which suppresses anti-tumor immune responses. Based on these data, we propose that blocking this pair of molecules will provide an opportunity to suppress melanoma growth. This project will attempt to elucidate the mechanisms through which this molecule pair regulates anti-tumor immune responses. In pursuing these studies, we hope to discover new ways to revive the tumor-killing function of our immune system and deliver new drug targets for the treatment of colon cancer and other types of cancers.
 S.Froehling
S.Froehling
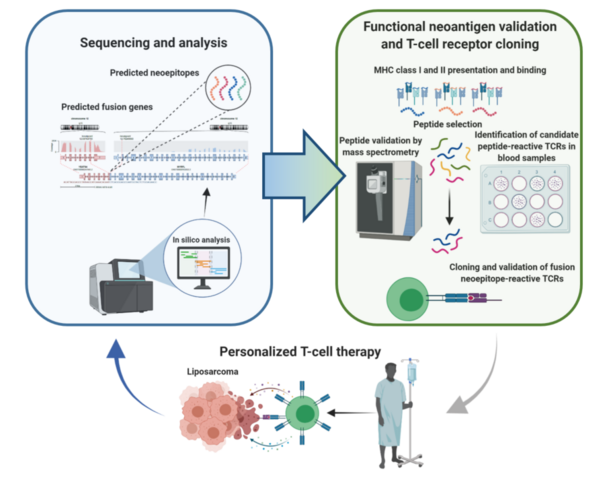
Liposarcomas are malignant soft-tissue tumors that are prone to local recurrence and distant metastasis. Medical therapy of patients with advanced liposarcoma, whose median survival is less than two years, represents a challenge since only a minority respond to conventional cytotoxic drugs and no approved targeted therapies exist. The most common histologic subtypes, well-differentiated and dedifferentiated liposarcoma (DDLS), are characterized by focal amplification of chromosome 12q, involving CDK4 and MDM2 genes. Using whole-exome/genome and RNA sequencing in DDLS patients within the DKFZ/NCT/DKTK MASTER program, we discovered that the structural changes on chromosome 12q generate open reading frames, leading to several hundred presumably transcribed and translated chimeric genes and neoantigens. Thus, DDLS may be accessible for immunotherapy. However, these events' randomness and diversity make the presence of a common neoantigen unlikely and instead favor individualized T-cell receptor (TCR)- or vaccination-based treatment strategies. The proposed project is based on preliminary data showing that the administration of individualized, fusion peptide-derived vaccines can induce measurable, peptide-specific T-cell responses in the blood of patients with DDLS. We now aim to systematically analyze gene fusion-derived neoepitopes and characterize patient-specific T-cell receptors in DDLS patients. This will lay the foundation for a scalable high-throughput pipeline to be used in future clinical trials with individualized TCR-transgenic T cells or personalized vaccines.
 C.Graf
C.Graf
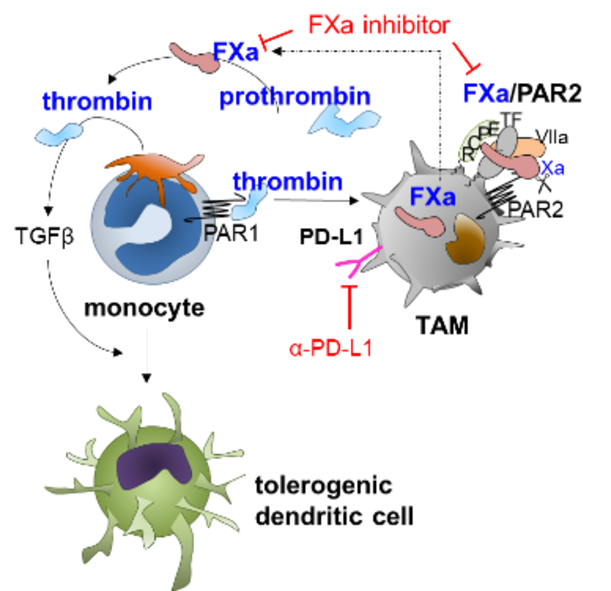
Current state of research: During malignancies systemic alteration of immune responses modulates blood coagulation, platelet-leukocyte interactions and pathological thrombus formation. This process is exacerbated by tumor cell specific expression of the coagulation proteins tissue factor, prothrombin and thrombopoietin leading to a hyperthrombotic state and thrombocytosis. Beyond regulation of blood clotting, coagulation proteases are causatively linked to tumor immune evasion as demonstrated for thrombin and activated factor X (FXa) in preclinical models 1,2. Thrombin acts on platelets to release the immunosuppressive and fibrosis inducing TGF-β, whereas FXa induces an immunosuppressive macrophage phenotype via autocrine FXa-protease-activated receptor 2 (PAR2) signaling, which converges with other innate immune signaling pathways to control dendritic cell phenotypes and to impair cytotoxic T-cell (CTL) responses. Both pathways are druggable with direct oral anticoagulants (DOACs), and their effects show synergism with checkpoint inhibitors. Complementary studies from tumor patients likewise showed increased monocyte-FX expression when compared to healthy controls indicating the translational relevance of our experimental studies. However, the detection of FXa in monocytes / TAM as a biomarker or the usage of DOACs as immune-modulators in tumor patients has not been clinically established.
Objectives: We aim to investigate the immunomodulatory function of coagulation signaling on the efficacy of clinically relevant immunotherapy in patients with or without FXa inhibitor co-medication. To this end, we aim to characterize the FX expression in peripheral blood monocytes and tumor-associated macrophages (TAM) of tumor patients at diagnosis and under treatment as a biomarker for tumor progression. Complementary studies in mice investigate the impact of platelet-monocyte aggregate abundance on monocyte phenotype and their abilities to differentiate into antigen presenting cells and T-cell priming. We posit that variable hyperthrombotic states and thrombopoetin expression of tumors modulate platelet counts and that tumor-educated platelets drive monocyte-differentiation into a tolerogenic state. As FXa activates prothrombin to thrombin, inhibition of FXa likewise inhibits thrombin generation. A better understanding of how coagulation signaling and different anticoagulants affects these processes will eventually allow for modifications of immunotherapies by combined usage with FXa inhibitors.
1. Graf C, Wilgenbus P, Pagel S, et al. Myeloid cell-synthesized coagulation factor X dampens antitumor immunity. Science immunology. 2019;4(39).
2. Metelli A, Wu BX, Riesenberg B, et al. Thrombin contributes to cancer immune evasion via proteolysis of platelet-bound GARP to activate LTGF-β. Science translational medicine. 2020;12(525).
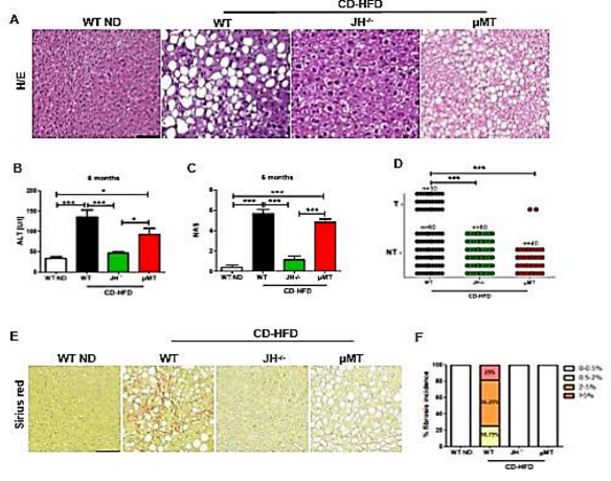 M.Heikenwaelder
M.Heikenwaelder
Obesity can lead to metabolic syndrome (e.g. abdominal obesity, insulin resistance) as well as hepatocellular carcinoma (HCC), which is the fastest growing cancer in the Western world, with strongly rising incidence in developing countries. Using long-term choline-deficient high-fat diet (CD-HFD) or Western Diet (WD), we could recapitulate the most important features of metabolic syndrome in humans - including the pathophysiology of non-alcoholic fatty liver disease (NAFLD), its severe pathology termed “non-alcoholic steatohepatitis (NASH)” and NASH-to-HCC transition. Besides, we have also established a model of alcoholic steatohepatitis (ASH) and NASH-ASH driven liver cancer, termed BASH (both alchoholic steatohepatitis forms). Today, the majority of people with fatty liver disease display BASH, due to a combination of high caloric diet, alcohol consumption and sedentary life style. Recently, B-cells have been considered as important mediators and players in innate and adaptive immune responses associated with metabolic diseases. Our preliminary data illuminate new mechanistic aspects on the role of B-cells in NASH development, liver fibrosis and liver cancer. Our data show that secretion of IgA drives FcR/FcR-signaling on inflammatory monocytes in the development of fibrosis, NASH and liver cancer. Recently, we could show that liver cancer patients with an underlying NASH/BASH do not respond to immunotherapy, but in contrast actually display adverse effects (e.g. reduced overall survival) (Dudek et al., Nature 2021; Pfister et al., Nature 2021). Thus, adjustment of existing therapies and better stratification is needed to optimize therapy outcome. B-cells might become an important target cell in this context - as besides immunoglobulin expression also B-cell derived antigen presentation contribute to hepatic dyslipidemia, NASH/BASH and liver cancer. To develop new, or adjusted immunotherapies for HCC patients (caused by NASH/BASH), we plan to use a B-cell directed immunotherapy and intend to target different B cell types or inhibit specific immunoglobulins in order to study how this therapy either prevents or cures HCC - e.g. in combination with an immunotherapy approach. We aim (ia) to perform Single cell RNA-Seq analysis of sorted hepatic and gastrointestinal CD45+ cells from different liver cancer mouse models (ib) to perform Single cell RNA-Seq analysis of CD45+ cells from human gastrointestinal and liver samples of NASH/BASH patients, liver cancer patients. We aim to (ii) to target newly identified, selected genes expressed in B-cells, and reinject genetically modified B-cells. We aim to (iii) to perform therapeutic B-cell- or immunoglobulin-A (IgA) depletion in mice with NASH/BASH and investigate liver cancer development in the presence or absence with mono- or combinatorial immunotherapy (anti-PDL1 and anti-VEGF). This study is lead by two PIs: Prof. Mathias Heikenwälder; Division Chronic Inflamaton and Cancer, German Cancer Research Center (DKFZ), Heidelberg; Prof. Ari Waisman; Institute for Molecular Medicine, University Medical Center of the Johannes Gutenberg University of Mainz; Focus Program Translational Neuroscience (FTN) Mainz; Research Center for Immunotherapy (FZI) Mainz, Mainz, Germany. The project greatly profits from the complementary expertise in immunology and cancer of the two individual PIs.
Pancreatic cancer is the currently most lethal tumor entity; mortality is close to incidence. In Western countries, it will become the second most frequent cause of cancer-related death behind lung cancer by 2030 due to the lack of means for early detection and therapeutic options. The DKFZ Division of Functional Genome Analysis has been working on pancreatic cancer for many years in close collaboration with the European Pancreas Center at Heidelberg University Hospital.
New data suggests that the humoral immune response and the T-cell reaction to pancreatic cancer may affect each other in a specific way. The project aims at understanding this interaction in sufficient detail to exert an influence on this process in a targeted manner. Thereby it may be possible to trigger a strong immune reaction to pancreatic cancer. Toward this end, the DKFZ group teams up with colleagues at the Immunotherapy Development Center of TRON, bringing together expertise from the areas of molecular biology, medical research and immuno-oncology. The project might lay the basis for a new approach of immunotherapy, which could improve the dismal prognosis of pancreatic cancer patients.
Helicobacter pylori (H. pylori), which infects about 50% of the world population, is associated with several gastropathies including chronic gastritis, peptic ulcer disease and mucosa-associated lymphoid tissue lymphoma. It causes gastric cancer in 1-3% infected humans. We hypothesize that the H. pylori-derived peptides are subject to CD8+ T-cell recognition and that H. pylori-specific CD8+ T cells can be isolated from the peripheral blood of patients with gastric cancer.
We will identify specific H. pylori antigens from literature, databases and our sequence-prediction algorithms using the genomic sequence and epitope prediction tools. Blood samples from gastric cancer patients will be screened for reactive T-cells using Elispot assays. In parallel, we will use NGS data from TCGA and GTEx as well as public mass-spec datasets to gain insight into the tumor specificity of the predicted antigens. We will use H. pylori-specific T-cells from selected H. pylori-reactive blood samples and stimulate them with selected H. pylori peptides. Next, the pool of antigen-specific reactive T-cells will be enriched followed by single-cell TCR sequencing. The subsequent bioinformatics analysis will allow the definition of H. pylori-specific TCR sequences. Alpha and beta chains will be cloned in a specific vector available in house to produce IVT RNA for each TCR chain, which will be tested for reactivity and specificity in primary T-cells and Jurkat cells, using a pool of autologous antigen-presenting cells preloaded with H. pylori-specific peptides.
The project will result in a proof-of-concept that antigens of bacterial origin can be presented on the cell surface of host cells in patients with gastric cancer. The detection of H. pylori-reactive T cells may give rise to a new class of immune-oncology biomarkers and interventional targets. These TCRs may be a starting point for an immunotherapy against H. pylori and gastric cancer.
 J.Faber
J.Faber
J.Faber
J.Faber
The treatment of pediatric patients can be defined as a success of medicine, with 82% of the patients under 15 years of age surviving their cancer for more than 15 years in Germany. However, cancer remains the leading cause of death by disease in children in developed countries and new therapy approaches are urgently needed.
Immunotherapy is changing the landscape of cancer treatment for adult patients, but is generally not used for the treatment of pediatric patients with solid tumors, with the exception of monoclonal antibodies against the ganglioside GD2 in neuroblastoma. Immunotherapy clinical trials in pediatric patients are still rare. This is due not only to the need to protect young patients from not well defined risk and toxicities, but also to our limited knowledge on the expression of molecular targets in pediatric tumors and in normal tissues of children. Pediatric cancers have indeed a unique etiology. While adult cancers usually occur as the result of a gradual accumulation of somatic mutations, pediatric cancers are rather considered as a developmental disease.
To accelerate the inclusion of pediatric patients in immunotherapy trials, in this project we will analyze the distribution of molecular targets in pediatric solid tumors associated with a poor prognosis, such as neuroblastoma, osteosarcoma, Ewing’s sarcoma and kidney tumors and in intracranial high-grade tumors, such as diffuse midline glioma and medulloblastoma. We will focus on plasma membrane targets that can be addressed by monoclonal antibodies and CAR-T cells in therapeutic protocols. Importantly, we will not only consider protein, but also lipid targets, which are not detectable by the pipelines generally used for targets identification. An important part of this project will be the establishment of a high quality set of normal pediatric tissues for testing the tumor specificity of the molecular targets.
The project is led by the Center for Pediatric and Adolescent Medicine of the University Medical Center (UM) Mainz (PI Prof. Dr. Jörg Faber and scientific coordination PD Dr. Claudia Paret) in collaboration with the Institute of Pathology (PI Prof. Wilfried Roth and Dr. Larissa Seidmann), the Institute of Neuropathology (PI PD Dr. Katrin Frauenknecht) of the UM Mainz, TRON gGmbH (PI Dr. Akilli-Oeztuerk) and the group of Lipid Pathobiochemistry of the DKFZ (PI PD Dr. Roger Sandhoff).
We anticipate that this project will facilitate the access of the pediatric population to immunotherapy clinical studies that are currently enrolling only adult patients and to Investigator Initiated Trials (IIT).
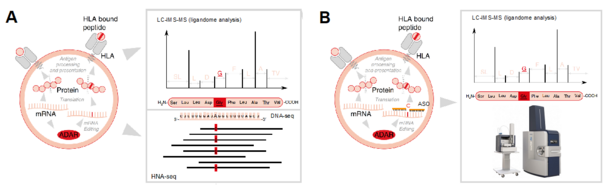 R.Pecori
R.Pecori
Successful immunotherapy depends on the potentiation of previously anergic, tumour-specific T cells, towards tumour cell killing. Current strategies include the reactivation of T cells via inhibition of their quiescent state through immune checkpoint inhibitors (ICIs). Despite their remarkable clinical successes, many patients do not respond to immunotherapy or develop therapeutic resistance. This aspect defines an urgent need for novel strategies that sensitize tumors to immunotherapy. Tumour mutational burden is the best predictor of responsiveness to immunotherapy, as its increase correlates with increased neoepitope/antigen formation, and therefore increased sensitivity to T cell attack. Therefore, specifically increasing neoepitope formation in cancer cells constitutes a promising strategy to sensitize tumors to immunotherapy. Neoantigens arise either from DNA mutations or are generated by post-transcriptional modifications at the RNA level (e.g., RNA splicing or retained introns caused by splicing errors). RNA editing, the most common post-transcriptional modification in mRNA, can induce non-synonymous substitutions generating neoantigens. The RNA editing enzyme adenosine deaminase acting on RNA 1 (ADAR1) deaminates adenosine to inosine (A-to-I) within double-stranded RNA (dsRNA) structures. In many tumours, ADAR1 is overexpressed and targets coding regions at the mRNA level, thus facilitating the generation of neoepitopes. Antisense oligonucleotides (ASOs) enable re-targeting of endogenous ADAR1 to coding regions leading to amino acid recoding. To pave the way towards novel, ADAR1-mediated tumor immunotherapy approaches, we will elucidate the efficiency of ADAR1-dependent neoantigen generation on the MHC ligandome level (A) and investigate approaches to specifically enhance neoepitope formation by using ASOs with the aim to increase the level of tumour “neoantigenicity” (B) and thus render the tumour sensitive to ICIs.
The project in enabled by a tight collaboration between PD Dr. Riccardo Pecori (DKFZ) and Prof Dr. Stefan Tenzer (University Medical Center Mainz / HI-TRON) merging their expertise in RNA base-editing and immunopeptidomics technologies.
 F.Vascotto
F.Vascotto
F.Vascotto
F.Vascotto
Immune checkpoints are key regulators in the control of anti-cancer immunity. In recent years, checkpoint inhibitors (CPI) have proven the possibility to mount immune responses against cancer inducing profound and durable clinical responses. Nevertheless, a large proportion of cancer patients unfortunately fall into relapse. Thus, there is an urgent need to deeper understand the underlying resistance mechanisms of cancers during checkpoint inhibitor therapies, in order to improve the clinical outcome of patients. Reinvigoration and release of suppressive mechanisms on intratumor CD8+ T cells, considered as the main anti-tumor effector cells, are the major aims of combination therapies. Based on our previous findings, we will investigate the tumor immune microenvironment (TiME) modeling in vivo and in vitro using murine tumor models and human primary tumors to study comprehensively the role of a novel axis involved in the control of T cell activity. Our aim is to generate evidence that this new axis in combination with checkpoint inhibitors can generate a new therapeutic approach for cancer patients.
Down-regulation or loss of HLA expression represents an important immune-escape mechanism and is a serious limitation for immunotherapy strategies relying on HLA-restricted effectors. HLA-independent anti-tumor T-cell responses have been observed in patients with HLA-negative disease. So far two of their target antigens have been identified, tyrosinase-related protein 2 (TRP2) and the GM-CSF receptor alpha chain (CSF2RA). The project aims at the preclinical testing of patient-derived, HLA-independent, anti-tumor T-cell receptors (TCR) against TRP2 and CSF2RA in mice to demonstrate their anti-tumor effects after adoptive transfer and to exclude relevant off-tumor and off-target effects applying in vivo tracking of TCR-transduced T cells and immunohistochemistry on tumor and normal tissues. In addition, it is planned to identify further HLA-independent, tumor-associated T-cell targets (A) via cDNA library-expression screening and (B) via reverse targeting of promising tumor-associated surface molecules. The project´s ultimate goal is to pave the way for clinical adoptive therapy studies with HLA-independent anti-tumor TCR and, finally, prove both relevance and therapeutic use of the naturally occurring HLA-independent anti-tumor T-cell repertoire. This would also support future efforts to augment such T-cell responses in patients and to circumvent adoptive transfer requirements.